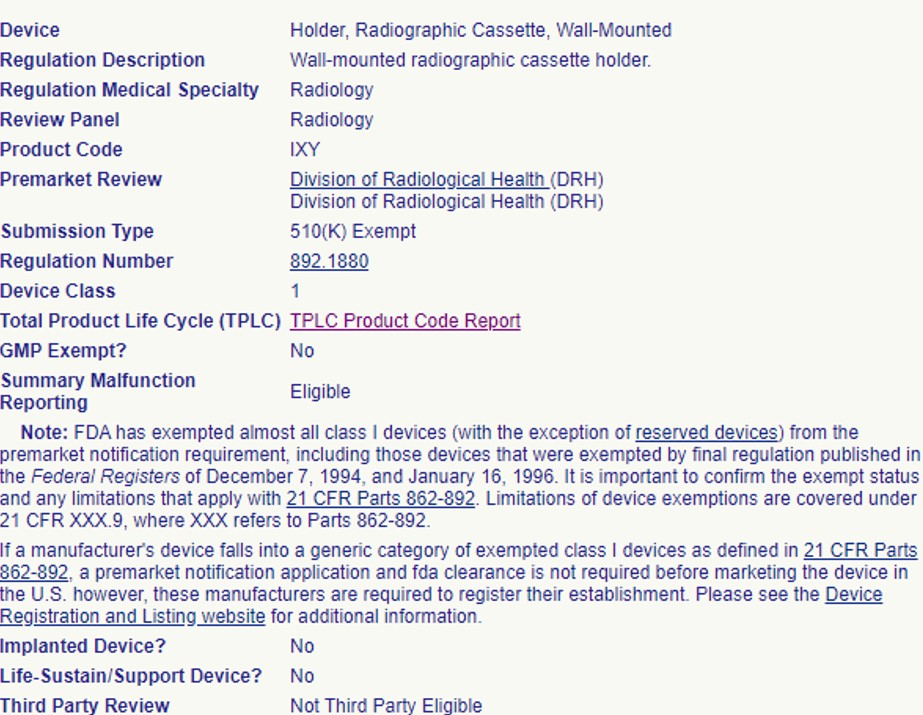
So, you have a medical device that you are ready to put on the market? You’ve identified a product code and the device is Class I or II and 510(k) exempt. With no 510(k) submission required, you are ready to register and list with the FDA and start selling your product, right? Not so fast!
It is important to confirm your device’s 510(k) exemption status and any limitations that may apply before you start selling the device. The FDA’s Federal Food, Drug, and Cosmetic Act describes “certain limitations” that would require a 510(k) submission for a device that appears to be 510(k) exempt on the surface.
The 510(k) exemption is limited “only to those devices that have existing or reasonably foreseeable characteristics of commercially distributed devices within that generic type”. If your device has unique characteristics and fits either of the following situations, the regulation says you are still required to submit a 510(k) prior to marketing:
- The device is intended for a use different from the intended use of a legally marketed device in that generic type of device; e.g., the device is intended for a different medical purpose, or the device is intended for lay use where the former intended use was by health care professionals only;
- The modified device operates using a different fundamental scientific technology than a legally marketed device in that generic type of device; e.g., a surgical instrument cuts tissue with a laser beam rather than with a sharpened metal blade, or an in vitro diagnostic device detects or identifies infectious agents by using deoxyribonucleic acid (DNA) probe or nucleic acid hybridization technology rather than culture or immunoassay technology;
While not listed in this article, there are even more considerations for the limitations that impact in vitro diagnostic devices.
CMD can help you navigate this scenario by identifying your product’s exemptions and the limitations to those exemptions. For example, being 510(k) exempt does not guarantee that the device is also GMP (Good Manufacturing Practices) exempt. (More on other common exemptions can be found here.)